Содержание
1. Причины развития болезни2. Классификация3. Симптомы4. Диагностика5. Лечение6. Прогноз развития болезни7. Когда срочно к врачу?8. FAQ
Аномалия Киари (мальформация Арнольда-Киари) – заболевание из группы врожденных пороков развития, при котором происходит опущение продолговатого мозга и части мозжечка, в норме расположенных в черепной коробке, в спинномозговой канал. Часто болезнь сочетается с дефектами костной системы – остеодистрофией, плоскостопием, шейным гиперлордозом, асимметрией лица, сращением первого шейного позвонка с затылочной костью. Некоторые формы сочетаются с недоразвитием структур мозга, гидроцефалией.
Диагностику проводят с помощью магнитно-резонансной томографии. Заболевание лечится хирургическими методами. Оперативное вмешательство проводится с целью декомпрессии сдавленных отделов мозга и обеспечения свободного движения спинномозговой жидкости. Операция позволяет улучшить состояние пациентов в 75% случаев.
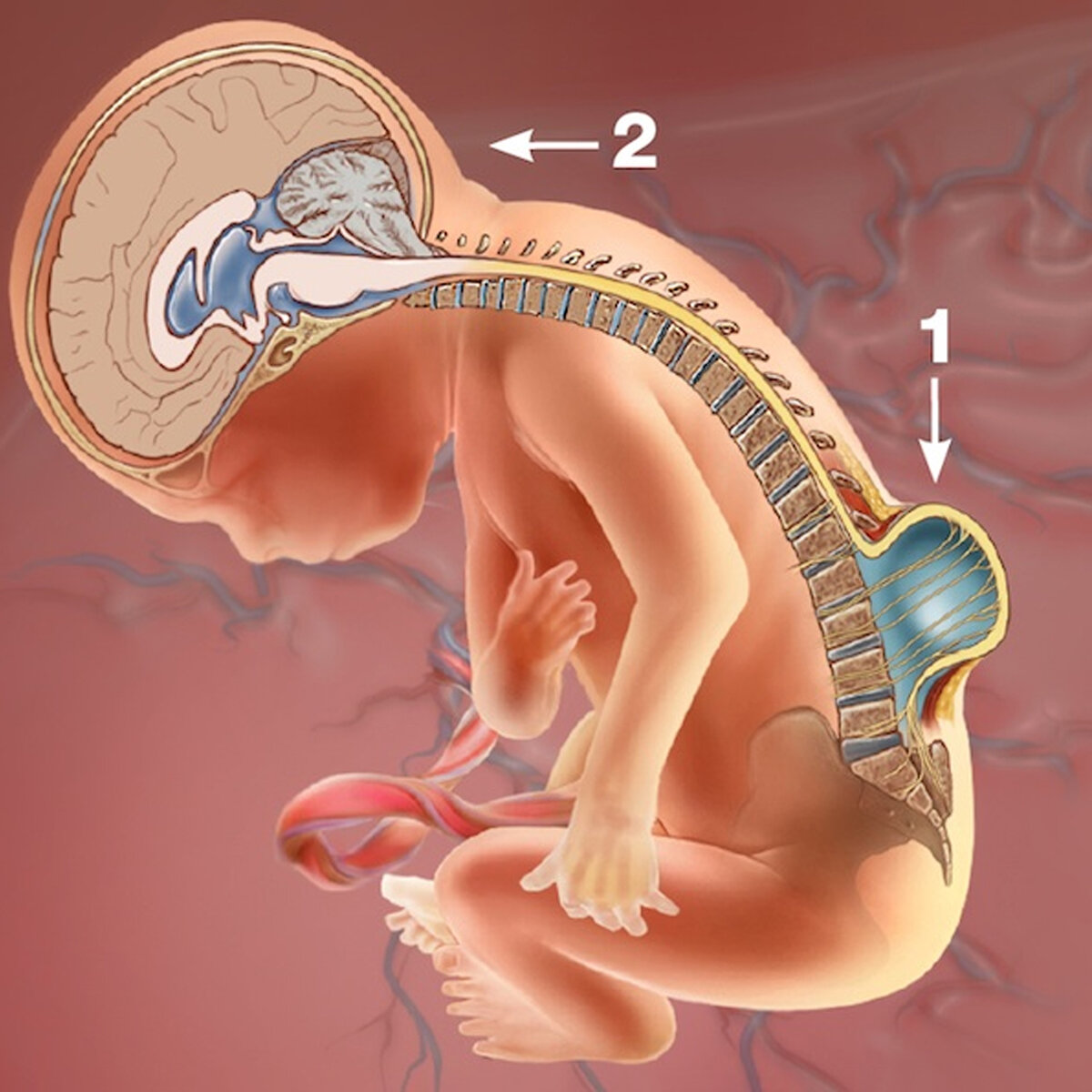
Спинной мозг переходит в головной на уровне большого затылочного отверстия. Все составляющие головного мозга (продолговатый, средний, промежуточный и конечный) в норме размещаются в черепной коробке, а спинной мозг – в канале позвоночника. При данной болезни нижняя часть головного мозга смещается вниз и выходит в спинномозговой канал. Происходит сдавление анатомических структур и нарушение циркуляции ликвора, что приводит к формированию гидроцефалии (избыточное скопление в желудочках мозга спинномозговой жидкости).
Синдром Арнольда-Киари относится к врожденным порокам развития нервной системы. Частота встречаемости заболевания – до 8 случаев на 100000 населения. Аномалия Киари, обнаруженная при рождении, часто несовместима с жизнью. У взрослых болезнь может быть диагностирована случайно при бессимптомном течении.
Причины развития болезни
Точные причины заболевания не установлены. Существует несколько теорий:
- неправильное развитие затылочной кости – недостаточный объем задней черепной ямки. Рост головного мозга и мозжечка сопровождается выходом из затылочного отверстия в спинномозговой канал;
- аномальное увеличение размеров головы – по мере роста становится недостаточно места в черепной коробке;
- наличие гидроцефалии с расширением мозговых желудочков и увеличением объема мозга;
- теория натяжения – укорочение концевой нити спинного мозга создает тягу, спинной мозг смещает головной по направлению к копчику, выводя его за пределы черепной коробки.
В развитии синдрома Арнольда-Киари играет роль наследственность – предрасположенность к тем или иным аномалиям мозга обусловлена генетически. Ухудшение состояния и прогрессирование симптомов болезни может вызвать травматическое повреждение позвоночника и спинного мозга, когда натяжение концевой нити усугубляется.
Классификация
Существует 4 типа аномалии Киари в зависимости от степени смещения мозговых структур и тяжести симптоматики:
- 1 тип – выпячивание миндалин мозжечка в позвоночный канал от 5 мм в виде грыжи, продолговатый мозг принимает уплощенную форму, сдавливая четвертый желудочек. Может протекать бессимптомно и выявляется у взрослых при диагностике других болезней;
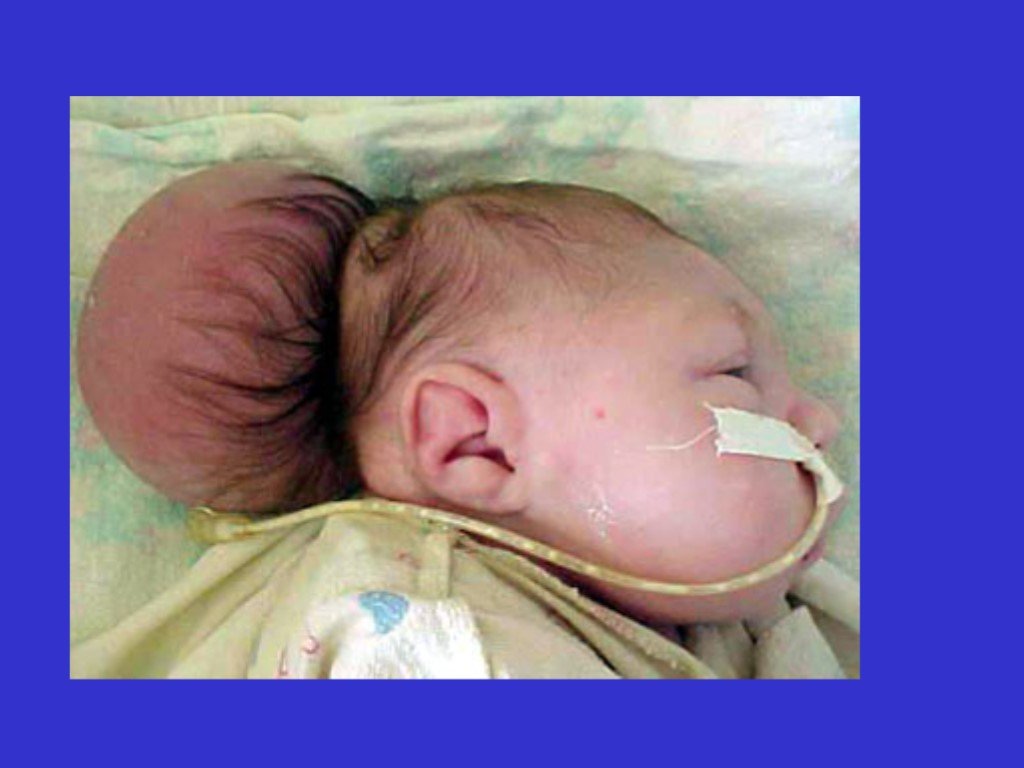
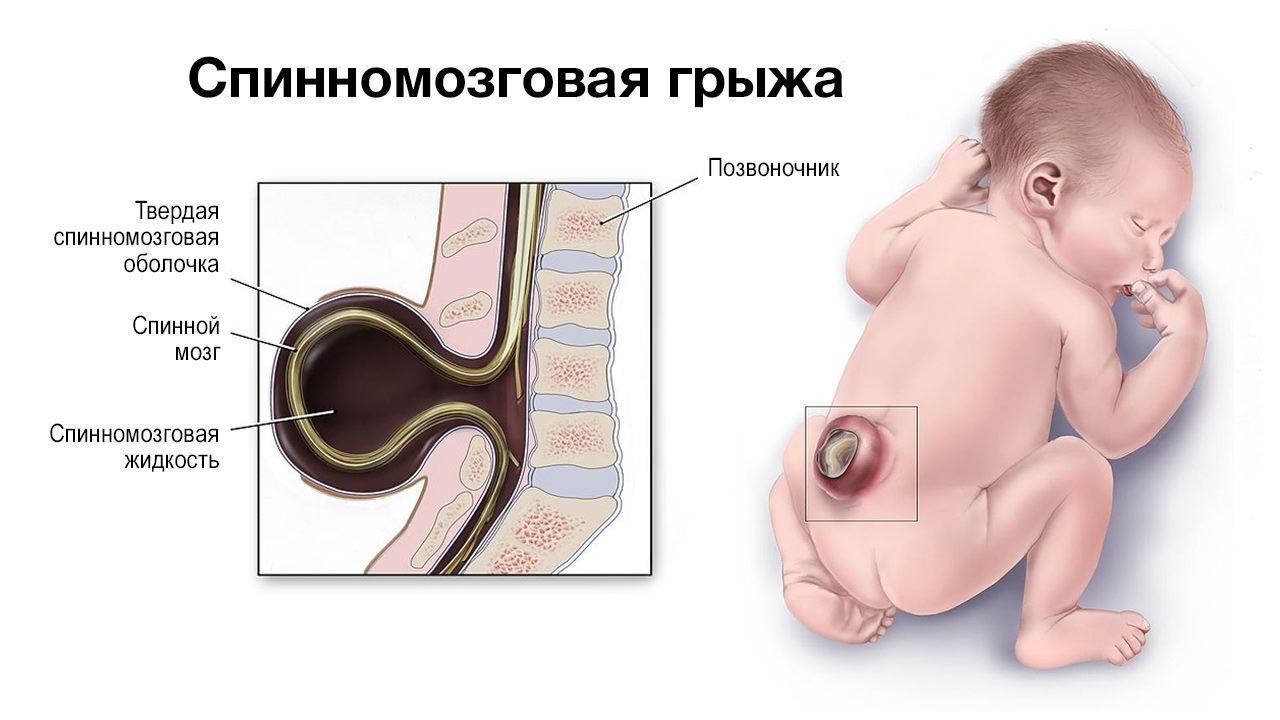
- 2 тип – значительное выпячивание в позвоночный канал средней части мозжечка, продолговатого мозга и четвертого желудочка. У пациента наблюдается миеломенингоцеле (грыжа спинного мозга), гидроцефалия, расщепление позвоночника в пояснично-крестцовой области;
- 3 тип – мозжечок и продолговатый мозг полностью опущены в позвоночный канал и образуют грыжевое выпячивание – энцефаломенингоцеле. Сочетается с расщеплением позвоночника в шейном отделе. Подобная форма несовместима с жизнью.
- Ранее выделяли 4 тип – резкое недоразвитие мозжечка без признаков его опущения. Сейчас эта патология относится к синдрому Денди-Уокера.
В последнее время выделяют болезнь 0 типа, характеризующуюся незначительным смещением или опущением мозжечка до уровня большого затылочного отверстия. Симптоматика обусловлена нарушением обращения цереброспинальной жидкости.
Симптомы
Клинические проявления болезни берут начало от следующих анатомических повреждений: сдавление ствола головного, спинного мозга, мозжечка и нарушение циркуляции цереброспинальной жидкости на уровне затылочного отверстия.
Клинические признаки болезни 1 типа начинаются в подростковом или зрелом возрасте. Для заболевания характерно:
- головная боль в задней части шеи, затылке, усиливающаяся при напряжении, чихании, кашле;
- головокружение, периодические обмороки, рвота, не связанная с приемами пищи;
- напряженность и боль в шейных мышцах;
- нестабильное зрение, снижение слуха, шум в ушах;
- нарушение жевания и глотания;
- осиплость голоса, затрудненное дыхание, обусловленное парезом гортани;
- парез конечностей;
- вегетативные нарушения – изменения артериального давления, ритма сердечных сокращений, остановка дыхания во сне, полидипсия, хроническая усталость, нарушения сна;
- нарушение походки, координации движений, дизартрия, нистагм.
Сирингомиелические кисты (полости в веществе спинного мозга) обуславливают следующие симптомы: расстройства чувствительности, онемение нижних конечностей, нарушение функции тазовых органов, мышечная слабость и гипотрофия. Размер и локализация кисты не всегда определяет силу проявления симптоматики.
Аномалия Киари 2 типа наблюдается с младенческого возраста и проявляется следующим образом:
- внезапные кратковременные остановки дыхания (апноэ), шумное дыхание (стридор), слабость голосовых связок (парез гортани);
- нарушения глотания с перемещением пищи в носовую полость;
- цианоз (посинение кожных покровов) во время кормления;
- нистагм;
- слабость и спазмы конечностей, тетраплегия (паралич четырех конечностей).
Болезнь 3 типа встречается редко, ее проявления аналогичны 2 типу, однако выражены сильнее и приводят к быстрой смерти новорожденного.
Диагностика
Диагностикой болезни занимается врач-невролог. Первичный осмотр и опрос пациента, родителей может быть дополнен инструментальными методами исследования (электроэнцефалография, ультразвуковое исследование, реоэнцефалография). Методы позволяют определить значительное повышение давления цереброспинальной жидкости (гидроцефалию).
Основной метод диагностики аномалии Арнольда-Киари – МРТ головного мозга и шейного отдела. Исследование неинвазивно, безвредно и максимально информативно в изучении мягких тканей. Серия послойных снимков позволяет увидеть в разных ракурсах краниовертебральную зону и черепную ямку с содержащимися в них мозговыми структурами. Исследование позволяет с точностью до миллиметра определить степень нарушения нормального строения мозга и поставить диагноз. У детей до 7 лет магнитно-резонансная томография проводится с участием анестезиолога, так как необходим общий наркоз.
Для данного заболевания характерны следующие признаки на МРТ:
- смещение миндалин мозжечка ниже линии, соединяющей твердое небо и затылочное отверстие;
- сирингомиелическая киста;
- признаки гидроцефалии.
Рентгенодиагностика и компьютерная томография не дают необходимой четкости визуализации, поэтому не используется для диагностики аномалии Киари.
Лечение

Бессимптомное течение часто остается незамеченным в течение всей жизни, поэтому не требует лечения. Малосимптомные формы аномалии Киари 1 типа, сопровождающиеся напряжением и болью мышц шеи, затылка, головной болью, лечатся консервативными методами. Терапия направлена на устранение негативных симптомов. Невролог назначает противовоспалительные, анальгезирующие средства, миорелаксанты, а также физиотерапевтические методы.
Недостаточность консервативного лечения, нарастание неврологической симптоматики, ведущая к ухудшению состояния и инвалидизации больного, развитие осложнений в виде гидроцефалии и сирингомиелии требуют проведения хирургического вмешательства. Суть операции заключается в обеспечении краниовертебральной декомпрессии и устранении сирингомиелический кист.
Расширение затылочного отверстия осуществляется за счет удаления небольшого участка затылочной кости, частичной резекции шейных позвонков. Сдавление ствола мозга и мозжечка ликвидируется путем резекции миндалин (удаляется до 70% объема). Нормализация циркуляции ликвора достигается путем рассечения твердой мозговой оболочки, сшивания и разведения в стороны твердой и арахноидальной оболочек, устранением спаек сосудов и продолговатого мозга с мозговыми оболочками. Операция завершается укреплением задней черепной ямки и пластикой твердой мозговой оболочки.
Другие подходы к хирургическому лечению болезни предполагают рассечение концевой нити спинного мозга, что устраняет патологическое натяжение и облегчает симптомы заболевания. Иногда выполняются операции по шунтированию – обеспечение оттока ликвора через искусственно созданные пути.
Прогноз развития болезни
Прогноз заболевания зависит от типа и своевременности диагностики и лечения. Болезнь 1 типа может не беспокоить пациента на протяжении всей жизни, но в некоторых случаях наблюдается ухудшение симптоматики. Важно обратиться к неврологу при появлении первых симптомов.
Аномалия Киари 2 требует наблюдения невролога и хирургического вмешательства. 3 тип часто не поддается лечению и приводит к смерти. Прогноз после операции благоприятный – у всех больных наблюдается улучшение состояния и положительная динамика болезни.
Когда срочно к врачу?
Немедленно обратитесь за медицинской помощью (вызовите скорую), если у вас или вашего ребенка возникли следующие симптомы, которые могут указывать на осложнения аномалии Киари или острое состояние:
- Внезапное нарушение дыхания или глотания, поперхивание.
- Внезапная сильная слабость или онемение в руках и ногах.
- Нарушение координации, шаткость походки, невозможность стоять или ходить.
- Двоение в глазах, нистагм (непроизвольные движения глаз).
- Потеря сознания или обморок.
- Судороги.
- Внезапная сильная головная боль, особенно в затылке, усиливающаяся при кашле или чихании.
- У новорожденных: внезапные остановки дыхания, синюшность кожи во время кормления, шумное дыхание.
FAQ
Что такое аномалия Киари (мальформация Арнольда-Киари)?
Это врожденный порок развития, при котором структуры задней черепной ямки (миндалины мозжечка, иногда ствол мозга) смещаются вниз и выходят через большое затылочное отверстие в позвоночный канал. Это приводит к сдавлению нервных структур и нарушению циркуляции спинномозговой жидкости. Выделяют 4 типа, из которых наиболее распространен 1-й тип, часто проявляющийся у взрослых.
Каковы основные симптомы аномалии Киари?
Симптомы разнообразны и зависят от степени сдавления. Наиболее частые: головная боль в затылке и шее, усиливающаяся при кашле, чихании, натуживании; головокружение; нарушение координации и равновесия; нистагм; двоение в глазах; нарушение глотания и речи; слабость и онемение в руках и ногах; шум в ушах, снижение слуха.
Как диагностируют аномалию Киари?
Основной и самый точный метод диагностики — МРТ головного мозга и шейного отдела позвоночника. Исследование позволяет увидеть степень опущения миндалин мозжечка, наличие сирингомиелической кисты (полости в спинном мозге), гидроцефалии и других сопутствующих аномалий. КТ и рентген менее информативны.
Как лечат аномалию Киари?
При бессимптомном течении лечения не требуется, показано наблюдение. При наличии симптомов, не снижающих качество жизни, возможно консервативное лечение (обезболивающие, миорелаксанты). При выраженных неврологических нарушениях, прогрессировании симптомов или развитии осложнений (сирингомиелия, гидроцефалия) проводится хирургическое лечение — декомпрессия задней черепной ямки для устранения сдавления.
Каков прогноз при аномалии Киари?
Прогноз зависит от типа и тяжести. Многие люди с 1-м типом живут бессимптомно всю жизнь. При своевременном хирургическом лечении прогноз благоприятный, у большинства пациентов симптомы уменьшаются или исчезают. 2-й и 3-й типы, проявляющиеся в детстве, имеют более серьезный прогноз и требуют раннего вмешательства.
Используемая и рекомендуемая литература
- PubMed: Langridge B., Phillips E., Choi D. Chiari Malformation Type 1: A Systematic Review of Natural History and Conservative Management. World Neurosurg. 2017;104:213–219. doi:10.1016/j.wneu.2017.04.082. Ссылка: https://pubmed.ncbi.nlm.nih.gov/28435116/ PubMed/
- CDC / WHO (совместное руководство): Birth Defects Surveillance: Quick Reference Handbook of Selected Congenital Anomalies and Infections. Geneva: WHO; 2020. ISBN 978-92-4-001541-8. Ссылка (PDF): https://archive.cdc.gov/www_cdc_gov/ncbddd/birthdefects/surveillancemanual/resource-library/quick-reference-handbook-P.pdf (год публикации: 2020).
- PubMed (гайдлайны CNS): Pattisapu J.V., Ackerman L.L., Infinger L.K., et al. Congress of Neurological Surgeons… Neurosurgery. 2023;93(4):731–735. Ссылка: https://pubmed.ncbi.nlm.nih.gov/37646504//
- Вахарловский В.Г., Горбунова В.Н. Клиническая генетика. Изд-во СПбГПМА, 2010.
- Можаев C.B. Особенности патогенеза, клиники и диагностики аномалии Киари 1 типа // Нейрохирургия. 2012.
- Шабалов Н.П. Неонатология. М.: ГЭОТАР-Медиа, 2016.