Содержание
1. Причины2. Клинические варианты патологии3. Диагностика4. Лечение и прогноз5. Когда срочно к врачу?6. FAQ
Болезнь Мачадо-Джозефа — довольно редкое неврологическое расстройство, проявляющееся в отсутствии мышечного контроля, что со временем при ненадлежащем лечении приводит к постепенному вырождению заднего мозга и гибели пациента. Диагностика осуществляется посредством оценки клинических проявлений патологии как у самого больного, так и у его ближних родственников, проведении КТ и МРТ пациенту, а также оценки копии триплета ЦАГ.
К сожалению, для постановки правильного диагноза обойтись исключительно визитом к врачу не получится, так как только комплексная диагностика может выявить заболевание даже на начальной стадии, что предоставит возможность специалисту назначить своевременную терапию.

Заболевание было впервые описано только в 70 гг. ХХ века. До этого периода в медицинской литературе нет никаких сведений о данной болезни. По предположениям ученых, патология впервые появилась у жителей Азорских островов, именно поэтому данное заболевание носит еще одно название — «азорская болезнь». Являясь самой распространенной формой мозжечковой атаксии, в наше время заболевание распространено повсеместно — случаи патологии были зарегистрированы в целом ряде стран.
Синдром Мачадо-Джозефа может проявить первые признаки у людей с 10 до 70 лет — довольно широкий возрастной диапазон, во время которого заболевание проявляет себя. Вследствие мультисистемного поражения спинальных и церебральных структур, проявления патологического процесса могут быть самыми разнообразными, что очень часто становится причиной постановки неправильного диагноза.
В зависимости от выраженности синдромов и характерных жалоб пациентов, принято различать три типа патологии. Множество форм проявлений очень сильно затрудняет диагностические мероприятия. Диагностика может быть осуществлена при совместной работе генетиков и неврологов. Только комплексная диагностика, проведенная при помощи новейших диагностических методик, которая будет оцениваться различными специалистами, способна предоставить максимально развернутую информацию для установки верного диагноза.
Причины

В тот период, когда патология была впервые обнаружена, специалисты не могли дать однозначного ответа на вопрос о том, каковы причины ее возникновения. Даже несмотря на то, что было выдвинуто множество предположений, ни одно из них не нашло своего научного подтверждения.
Однако, с течением времени и по мере развития ДНК-исследований стало ясно, что главными причинами болезни выступает мутация генов, передаваемая по наследству аутосомно-доминантным путем.
Таким образом удалось выяснить, что заболевание имеет наследственный характер. Отклонения от нормы наблюдаются в 14 хромосоме и сводятся к экспансии комбинации «цитозин-аденин-гуанин». Количество повторов триплета ЦАГ очень сильно различается, в среднем показатели держатся в диапазоне от 62 до 84. Для сравнения: в норме эта цифра не должна быть выше 37. Чем данный показатель выше, тем раньше возникает болезнь у человека.
У больных возникает апоптоз нейронов в коре головного мозга, дегенеративные процессы красного и зубчатого ядер, моторных ядер ЧМ нервов. Поражение зон мозга приводит к возникновению целого ряда признаков, которые по мере развития патологии и отсутствия соответствующего лечения начинают тревожить больного. Специалисты сходятся во мнении, что от момента возникновения патологии до проявления первых ее симптомов проходит от 3 до 6 месяцев, но многое зависит также от индивидуальных особенностей организма – у одних больных патология может проявиться значительно раньше, у других – позже.
Клинические варианты патологии

Патология первого типа возникает в возрастном диапазоне 10-30 лет. У больных ярко выражены экстрапирамидные и пирамидные симптомы развития патологии. Пирамидный синдром возникает вместе со спастическим парапарезом, постепенно появляется значительная слабость в верхних конечностях, парез мышц глотки, глазодвигательных нервов, в результате чего человек теряет способность передвигать глазными яблоками.
Со временем развиваются патологические рефлексы, клонус стоп. Больные начинают испытывать трудности при передвижении - походка становится неуклюжей, скованной, при этом человек ходит с широко расставленными ногами. Очень часто такие пациенты сильно шатаются во время ходьбы. Нередко у больных смещаются глазные яблоки вперед, возникают значительные фасцикулярные сокращения языка, мимических мышц, глазных век. Имеют место непроизвольные движения глаз высокой частоты.
Патология второго типа возникает чаще всего с 20 до 40 лет. Симптомы заболевания проявляются в мозжечковой атаксии: возникает дизартрия, больной не способен сохранять равновесие, появляется абазия. Характерной комбинации мозжечковых и пирамидных признаков, но второй тип имеет некую схожесть с первым типом болезни.
Патология третьего типа выражается в комбинации амиотрофий и мозжечковой атаксии. Данный тип заболевания возникает довольно поздно – как правило, только после 40 лет. У больных появляется мышечная атрофия, которая проявляется сильной слабостью и полной потерей сухожильных рефлексов. Особенностью данного типа болезни является наличие всех видов расстройств чувствительности по дистальному типу. Дегенеративные изменения в кортикоспинальных трактах и поражение экстрапирамидной системы не наблюдается.
Диагностика

Диагностика болезни осложняется тем, что ей свойственна огромная вариабельность повторов триплета ЦАГ, в результате чего болезнь проявляется в самых разнообразных симптомах даже в том случае, если возникает у членов одной семьи. Очень часто болезнь поражает разные поколения близких родственников, причем не обязательно проявляется в одном и том же типе.
Известны случаи, когда только одно из проявлений позволяло заподозрить наличие болезни у разных людей, имеющих между собой родственную связь. Одним из важнейших моментов в диагностике заболевания является консультация генетика, который осуществляет исследование генеалогического древа, учитывая большое количество родственников, а также проводя ДНК-исследование.
Немаловажным является и посещение невролога. Врач проведет первичный осмотр, по результатам которого выявит отличия болезни от других заболеваний.
К сожалению, в некоторых случаях осмотр у невролога не позволяет выявить характерные признаки заболевания из-за стертой клинической картины, поэтому врач может только заподозрить патологию, но поставить такой диагноз без более подробного обследования не получится. Пациентов с подозрениями на данный недуг в обязательном порядке направляют на КТ и МРТ головного мозга, а после чего врач-рентгенолог определит наличие изменений в ЦНС. Важно понимать, что только обследование, проведенное на современных аппаратах, предоставит возможность врачам дать оценку возникшим изменениям в структуре головного мозга, на основании которых будет поставлен правильный диагноз.
Лечение и прогноз
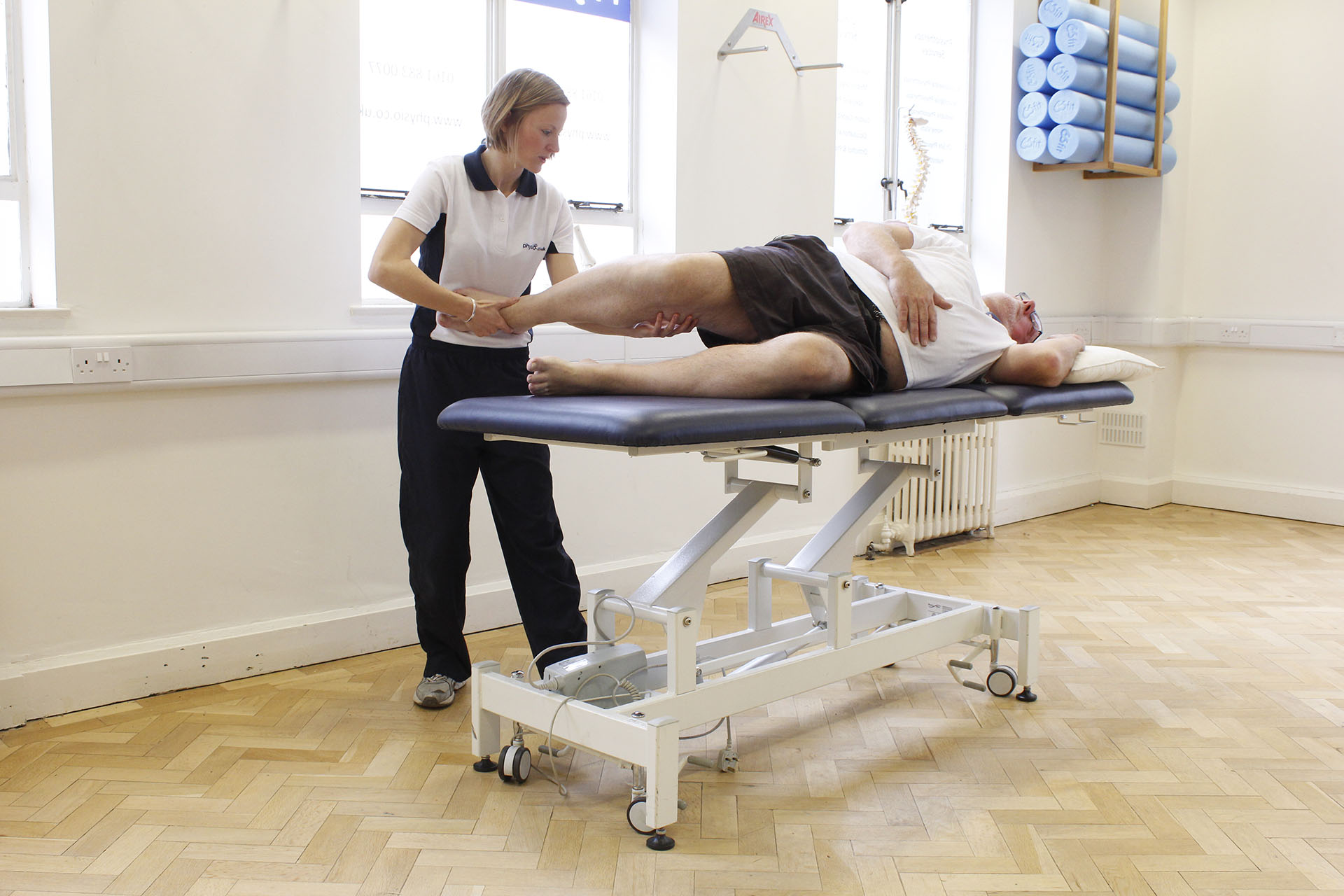
К сожалению, в настоящее время заболевание считается неизлечимым и никакого специального лечения, направленного на остановку дегенеративных процессов пока разработать не удалось. Поэтому все мероприятия направлены исключительно на снятие симптомов и улучшение качества жизни больного. При своевременно начатом лечении, которое было подобрано верно, а также соблюдении всех рекомендаций врачей, больные получают все шансы на замедление дегенеративных процессов, происходящих на фоне протекания болезни.
Однако, полного выздоровления не наступает даже при правильной терапевтической тактике. Если присутствует синдром паркинсонизма, назначают агонисты дофамина, иногда пациентам показано применение амантадина. С целью снятия спастики выписываются лекарственные препараты с миорелаксирующим действием.
Как уже говорилось выше, почти всегда даже хорошее лечение не сможет остановить дальнейшее развитие заболевания, поэтому с течением времени у больных появляются новые симптомы патологии, что в конечном итоге приводит к смерти. Согласно официальным статистическим данным, после выявления заболевания до гибели больного проходит от 10 до 20 лет.
Продолжительность жизни имеет прямую связь с типом заболевания. Неблагоприятный прогноз имеет первый тип Масадо-Джозефа, больные с таким диагнозом, как правило, не живут дольше 10 лет после начала прогрессирования патологии.
Профилактические мероприятия сводятся к проведению пренатальной диагностики, а также в получении консультации у генетиков. Если у ближайших родственников был диагностирован синдром Мачадо-Джозефа, важно со всей ответственностью подходить к планированию и рождению ребенка, так как генная мутация может передаться по наследству. Многие генетики сходятся во мнении, что очень важно не допускать рождение ребенка, у которого имеется характерная генетическая мутация.
Когда срочно к врачу?
Немедленно обратитесь за медицинской помощью, если у вас или ваших близких появились следующие симптомы:
- Внезапное нарушение координации, шаткость походки, невозможность стоять или ходить.
- Внезапное двоение в глазах, нарушение движения глаз.
- Нарушение речи (невнятность, смазанность).
- Затруднение глотания, поперхивание.
- Внезапная слабость в руках или ногах.
FAQ
Что такое болезнь Мачадо-Джозефа (спиноцеребеллярная атаксия 3 типа) простыми словами?
Это редкое наследственное заболевание нервной системы, при котором постепенно поражаются отделы мозга, отвечающие за координацию движений, равновесие, речь и движения глаз. Болезнь прогрессирует и приводит к тяжелой инвалидизации. Относится к группе спиноцеребеллярных атаксий.
Каковы основные симптомы болезни?
Симптомы разнообразны и зависят от типа. Основные проявления: нарушение координации (атаксия), неустойчивость при ходьбе, дизартрия (нарушение речи), нистагм (непроизвольные движения глаз), двоение в глазах, мышечная слабость, спастичность, а также симптомы паркинсонизма (скованность, тремор).
Как передается болезнь Мачадо-Джозефа?
Заболевание наследуется по аутосомно-доминантному типу. Это значит, что для развития болезни достаточно одной мутантной копии гена, полученной от одного из родителей. Риск передачи болезни ребенку составляет 50%.
Как диагностируют болезнь Мачадо-Джозефа?
Диагноз ставится на основе клинической картины, семейного анамнеза и подтверждается генетическим анализом (поиск мутации в гене ATXN3). МРТ головного мозга может показать атрофию мозжечка и ствола мозга, что помогает в диагностике и исключении других заболеваний.
Существует ли лечение болезни Мачадо-Джозефа?
На сегодняшний день болезнь неизлечима. Терапия направлена на облегчение симптомов и улучшение качества жизни. Она включает лекарства для уменьшения спастичности, тремора, а также физиотерапию, логопедическую помощь и реабилитацию. Продолжительность жизни после появления симптомов варьирует, но в среднем составляет 10-20 лет.
Используемая литература
- Ribeiro RFN, Pereira D, Lopes SM, et al. Circadian rhythms are disrupted in patients and preclinical models of Machado-Joseph disease. Brain. 2025 Jul 22. doi:10.1093/brain/awaf199. PMID:40692435. PubMed.
- do Carmo Costa M. Recent therapeutic prospects for Machado-Joseph disease. Current Opinion in Neurology. 2020;33(4):519–526. doi:10.1097/WCO.0000000000000832. PMID:32657894.
- Dantuma NP, Herzog LK. Machado-Joseph Disease: A Stress Combating Deubiquitylating Enzyme Changing Sides. Advances in Experimental Medicine and Biology. 2020;1233:237–260. doi:10.1007/978-3-030-38266-7_10. PMID:32274760.
- Paulson HL. Spinocerebellar Ataxia Type 3. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; updated 2020. URL: https://www.ncbi.nlm.nih.gov/books/NBK1196/ (публикация в сети; дата обновления — 2020). NCBI.
- Bettencourt C, Lima M. Machado-Joseph Disease: from first descriptions to new perspectives. Orphanet Journal of Rare Diseases. 2011;6:35. doi:10.1186/1750-1172-6-35. BioMed Central.
- MedlinePlus Genetics. Spinocerebellar ataxia type 3. 2019-02-01. URL: https://medlineplus.gov/genetics/condition/spinocerebellar-ataxia-type-3/ (публикация в сети; дата публикации — 2019-02-01). MedlinePlus.