Содержание
1. Общее понятие2. Полезные сведения3. Патогенез невральной амиотрофии Шарко-Мари-Тута4. В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?5. Варианты диагностики6. Способы лечения
Общее понятие
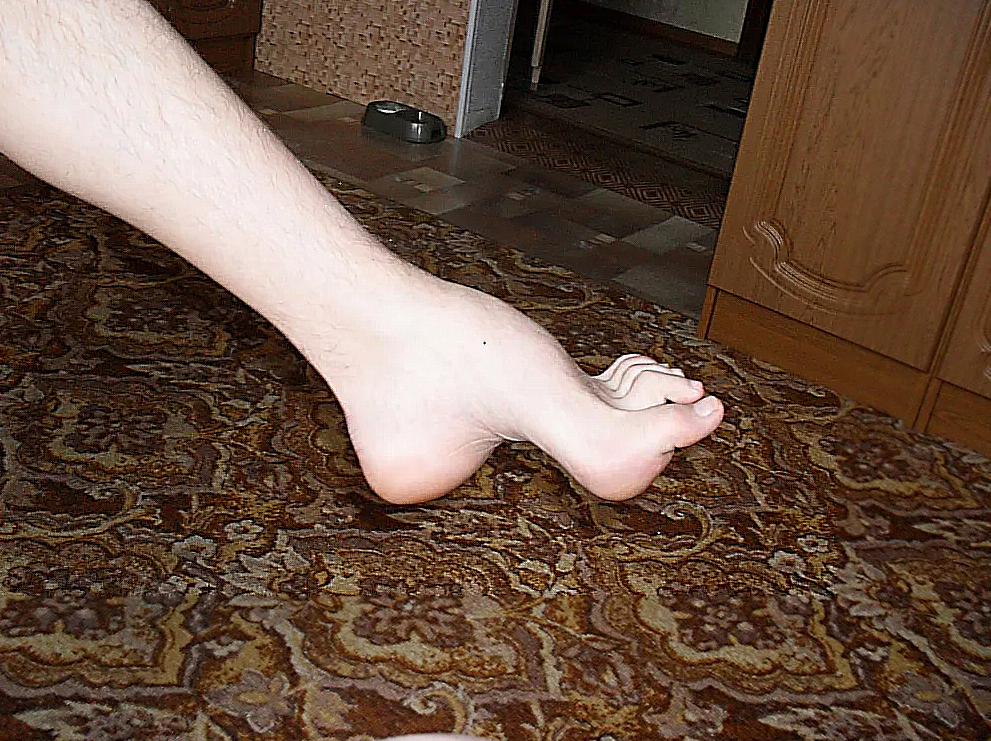
Невральная амиотрофия Шарко-Мари-Тута — хроническое заболевание организма, носящее наследственный тип образования. Характерным показателем является поражение нервной системы периферического отдела. Аномальные процессы проявляются в формировании и изменений структуры в мышечных зонах, например: уменьшение в размерах районов в начале нижних конечностей, а после — верхних. В комбинации с аномалией данного вида у больного может возникнуть гипестезия и снижение рефлекторной способности сухожилий, подергивания разных отделов мышц.
Врачи прибегают к различным вариантам обследования пациента с таким недугом, к ним относятся: электромиография, электронейрография, генетическое обследование, ДНК-проверка, биопсия нервов и мышечных участков. Радикальных приемов лечения — не существует, но имеются методики направленные на избавление человека от симптомов. Медики предписывают разнообразные витаминные комплексы, антихолинэстеразной способы, метаболические приемы, микроциркуляторные аналоги, ЛФК, физиотерапию и прочее.
Полезные сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) входит в список хронических заболеваний (полиневропатий) генетического характера, которые проявляются в повышенном прогрессировании. К ней относятся:
- Синдром Русси-Леви.
- Гипертрофическая невропатия Дежерина-Сотта.
- Болезнь Рефсума и другие более редкие патологические процессы.
Согласно среднестатистическим данным ученые заявляют, что практически 83% случая вызваны в результате наследственной предрасположенности человека. Неблагоприятные действия в теле возникают по большей мере у мужчин, женщины менее подвержены описываемому заболеванию. Ссылаясь на многочисленные сведения, невральная амиотрофия ШМТ диагностируется с частотой от двух до тридцати шести случаев на сто тысяч населения планеты. Обычно, она носит семейный характер. Однако у членов одной семьи симптоматика клиника может быть по-разному выражена. Вместе с этим врачи наблюдают и спорадические варианты ШМТ. Медицинские сотрудники отмечают четкую взаимосвязь с потерей координации по Фридрейху. У некоторых больных в разных ситуациях замечаются типичные симптомы одной или другой болезни. Иногда, с течением долгих лет, клиника одной аномалии может замениться симптомами другой.
Патогенез невральной амиотрофии Шарко-Мари-Тута
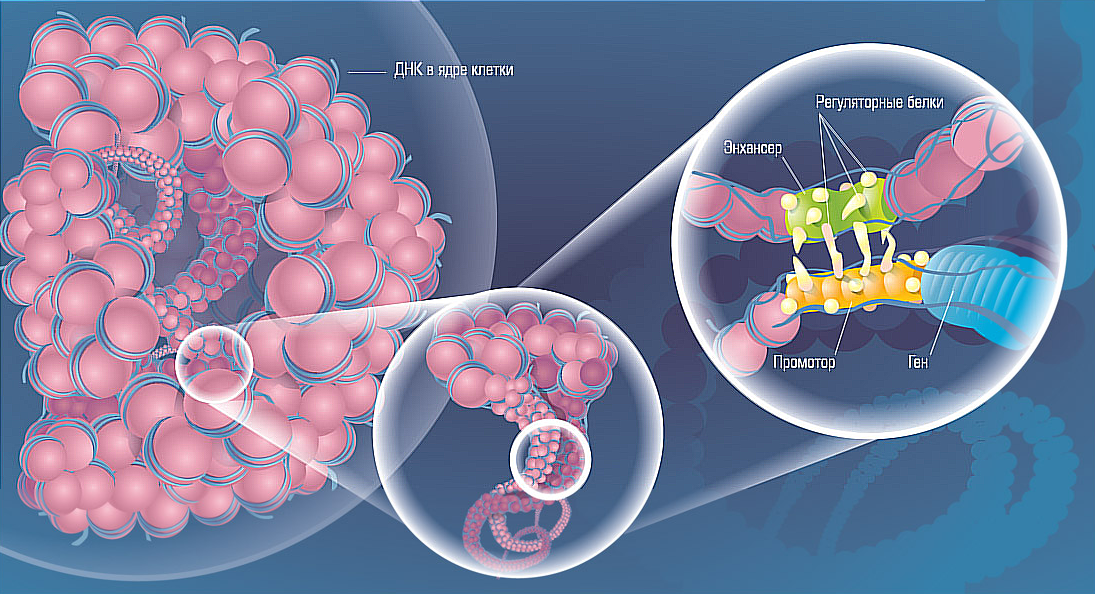
В медицине на данный момент не имеется точной и достоверной информации о происхождении и механизме возникновения невральной амиотрофии. Согласно проведенным экспериментам стало понятно только то, что почти у 75% пациентов с таким диагнозом, которые прошли генетическую консультацию, выделялось повторение определенной зоны 17-й хромосомы. Уже известно, что заболевание обладает несколькими группами, вызываемыми различными мутациями генов. Так, например, во время исследования больного с ШМТ, проявляющейся из-за мутации белка гена MFN2, начинается появление частиц митохондрий. В результате чего у человека происходит сбой в движении этих элементов по аксону.
Большое количество форм проявляются вследствие поражения миелинового наружного слоя волокон. Намного реже можно заметить формы с отклонением от нормы аксонов — объектов осевого направления, которые проходят в центральной части нервной структуры. Аномальные процессы способны повлиять на состояние передних и задних отростков спинного мозга, нейронов передних рогов, путей Голля и столбы Кларка. Повторно из-за проблем с функционированием периферического отдела, начинает образовываться мышечная недостаточность.
Разновидности и классификация
В неврологическом направлении медицины невральная амиотрофия Шарко-Мари-Тута разделена на две четких группы, которые клинически достаточно схожи между собой, но имеют список особенностей, позволяющих провести подобное разграничение:
- Невральная амиотрофия I типа — вызывает снижение скорости проведения нервного импульса.
- НА II типа — скорость подвергается пагубному влиянию в меньшей доли, проявляется отклонение нейрита.
В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
При описываемом заболевании у человека первым делом берет начало формирование схожих мышечных некрозов в нижних конечностях. Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
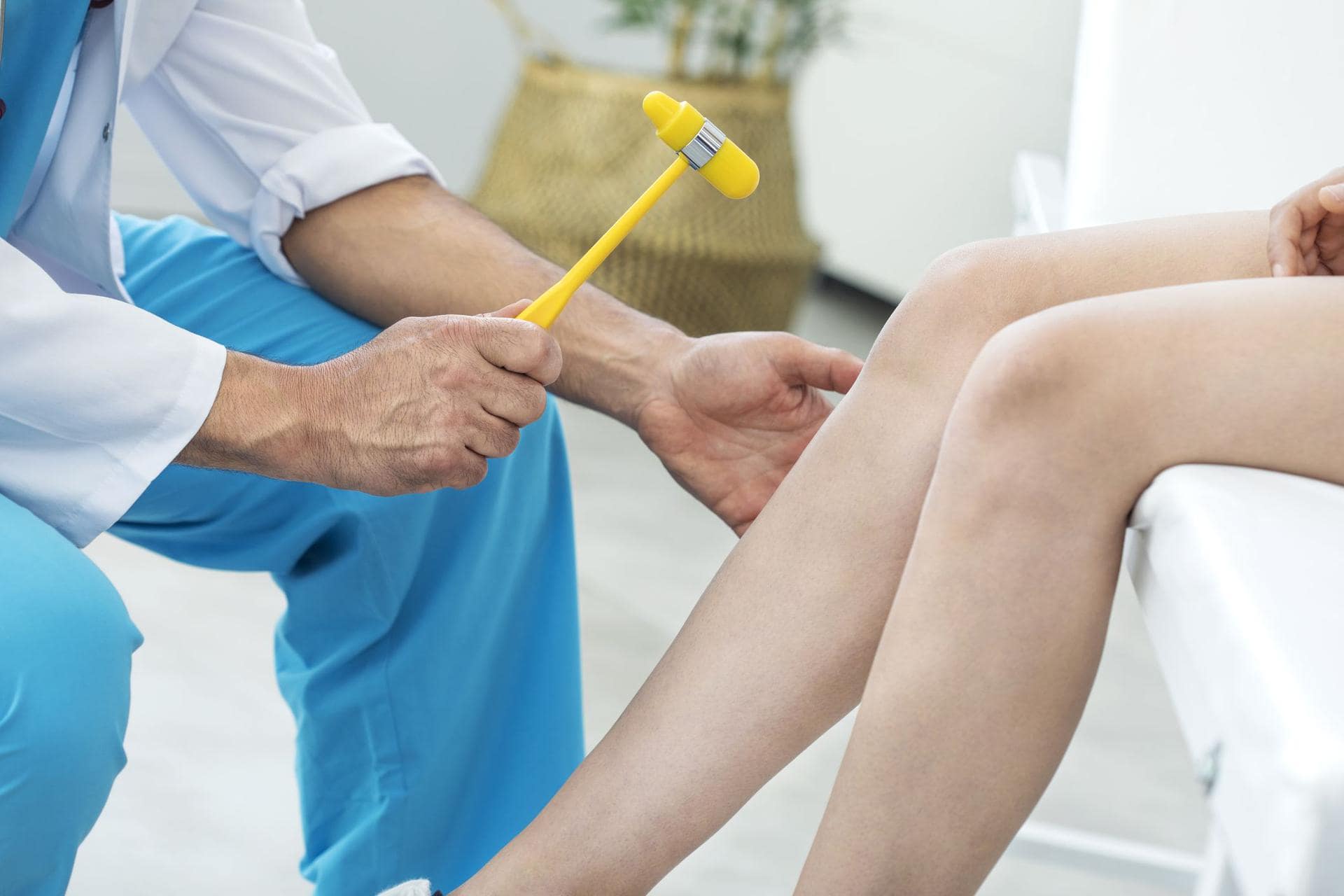
Типичным ранних звоночком ШМ, считается полное отсутствие ахилловых и коленных рефлексов сухожилий. В итоге у больного проявляется свисание свода, неспособность ходить на пятках и анормальная походка, которая схожа с лошадиной. Впоследствии пагубный процесс увеличивает степень прогрессирования дальше и затрагивает мышцы и сгибатели. Максимальная уровень омертвения может привести к полной деформации ног с высоким сводом, схожу на тип стопы Фридрейха. Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Стоит знать, что пагубные поражения никогда не поражают мышцы шейного периметра, туловища и плечевого района. Помимо вышеперечисленных симптомов, у больного могут появиться следующие проблемы:
- Слабое подергивание.
- Гипертрофия компенсаторной природы мышечных областей.
- Сенсорные нарушения.
- Возможность появления цианоза и отечности на кожном покрове.
Для ШМТ типично медленное развитие симптоматики. Период развития процесса уменьшения в размерах ног и рук может занять до десяти лет. Даже при таких серьёзных деформациях тела больной долгое время способен сохранять работоспособность и нормально выполнять различные бытовые обязанности. Ускорителями симптомов могут стать следующие факторы:
- Попадание в тело инфекционных агентов.
- Долгое пребывание на холоде.
- Травмирования головы.
- Повреждения спины и спинного мозга.
- Нехватка витаминов в организме.
Варианты диагностики
Медицинские работники для диагностирования НА Шарко-Мари-Тута опираются на такие проявления, как:
- Возраст, в котором появились первые признаки недуга.
- Стандартная клиническая картина.
- Схожий характер разрушения организма.
- Медленная степень развития атрофий.
- Увеличение опасности симптомов.
Во время посещения кабинета специалиста, человеку проведут полноценный осмотр тела, чтобы выявить наличие мышечной слабости в стопах и голенях, изменения, отсутствие или серьезное снижение уровня рефлекторных способностей ахиллова и коленного отдела, гипестезию. Для того чтобы отличить ШМТ от других нервно-мышечных расстройств, врачи составляют ряд обследований, которые потребуется пройти инфицированному. К ним относятся:
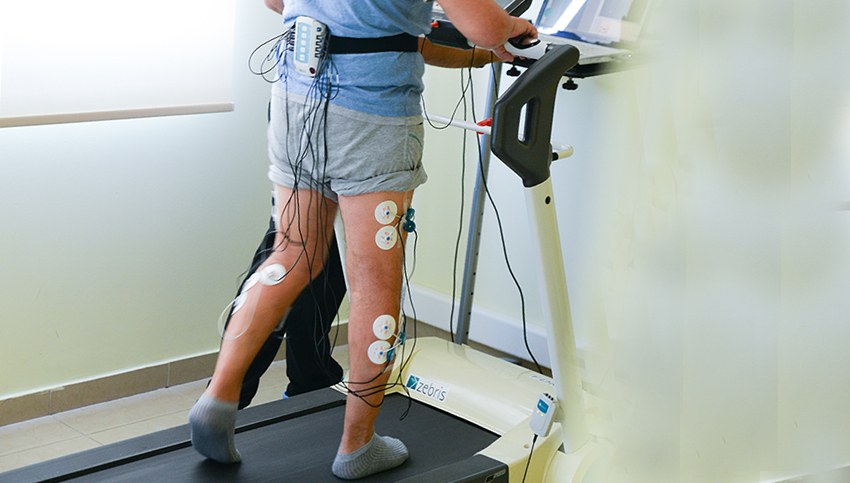
- Электромиографическое исследование.
- Электронейрография.
- МРТ конечностей.
- Анализ кровеносных телец на сахар, гормональную норму и наркотические вещества.
- Проведение консультации у специалиста в области генетики.
- ДНК-оценка или секвенирование генома (последний вариант очень дорогостоящий для широкого применения).
- Биопсия.
Способы лечения
В настоящее время медики и ученые всего мира не смогли разработать радикальные варианты избавления от описываемого заболевания. Поэтому для снижения неприятных ощущений у людей с ШМТ применяется симптоматические методики. Больному делают курсы внутримышечного введения витаминов группы В и Е. Для повышения состояния трофики используется АТФ и прочие вещества. Пациентам предписываются ингибиторные препараты холинэстеразы, фармакологические средства для повышения микроциркуляции, особый вид кислот и прочие лекарства. Помимо разных медикаментов больным рекомендуется проходить физтерапевтические процедуры. К ним относятся:
- Электрофорез — при которой организм человека подвергается влиянию электрических сигналов.
- Амплипульстерапия.
- Диадинамотерапия
- Грязелечение.
- Ультразвуковое лечение.
- Оксигенобаротерапия.
- Водолечение в сероводородных, сульфидных, хвойных, радоновых ваннах.
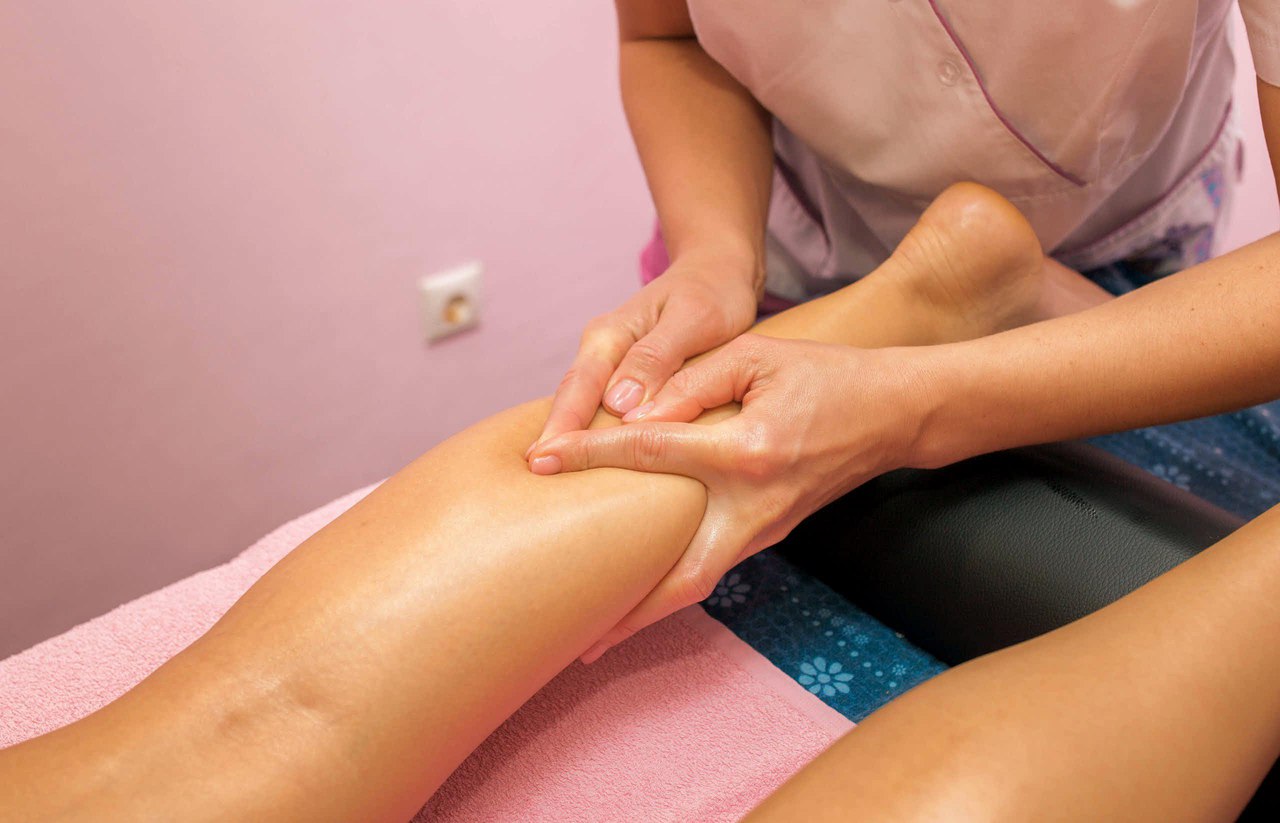
Большое значение для выздоровления человека носит сохранение регулярных двигательных нагрузок, предотвращение формирования изменений и прочих аномалий.
Именно для такого используется ЛФК и массажные сеансы.
При острой необходимости врач-ортопед может предписывать разные ортопедические приемы.
Используемая и рекомендуемая литература
- PubMed: Pareyson D., Marchesi C. Diagnosis, natural history, and management of CMT. 2009. https://pubmed.ncbi.nlm.nih.gov/19539237/.
- CDC (PHGKB): Charcot-Marie-Tooth disease — Rare Diseases (агрегатор по публикациям и ресурсам CDC/NLM). https://phgkb.cdc.gov/PHGKB/TopicDetail?topic=Charcot%20Marie-Tooth%20Disease&language=English.
- ВОЗ (ICD-11): Charcot-Marie-Tooth disease — страница классификации. https://icd.who.int/dev11/f/en#/http://id.who.int/icd/entity/466800293
- Глущенко Е.В. Клинико-генетическая характеристика наследственной невропатии Шарко-Мари-Тута (на примере Красноярского края): Автореф. дис. ... канд. мед. наук. Красноярск; 2011.
- Шнайдер Н.А. Оценка качества жизни больных с наследственной невропатией Шарко-Мари-Тута в Красноярском крае. Бюллетень Сибирской медицины. 2011.
- Епифанов В.А. Медицинская реабилитация. Руководство для врачей. М.: МЕДпресс-информ, 2005.
- Bird T.D. Charcot-Marie-Tooth Hereditary Neuropathy Overview. GeneReviews® [Интернет]. Seattle (WA): University of Washington, Seattle; 1998–2025. Обновлено 23.01.2025. URL: https://www.ncbi.nlm.nih.gov/books/NBK1358/ NCBI.
- Pareyson D., Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurology. 2009;8(7):654–667.
- Rossor A.M., Polke J.M., Houlden H., Reilly M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nature Reviews Neurology. 2013;9(10):562–571.
- Murphy S.M., Laura M., Fawcett K., et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. Journal of Neurology, Neurosurgery & Psychiatry. 2012;83(7):706–710.
- Laurá M., Pipis M., Rossor A.M., Reilly M.M. Charcot-Marie-Tooth disease and related disorders: an evolving landscape. Current Opinion in Neurology. 2019;32(5):641–650.
- Sman A.D., Hackett D., Fiatarone Singh M., et al. Systematic review of exercise for Charcot-Marie-Tooth disease. Journal of the Peripheral Nervous System. 2015;20(4):347–362.
- Yiu E.M., Bray P., Baets J., et al. Clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery & Psychiatry. 2022;93(5):530–538.
- Pfeffer G.B., Gonzalez T., Brodsky J., et al. A Consensus Statement on the Surgical Treatment of Charcot-Marie-Tooth Disease. Foot & Ankle International. 2020;41(7):870–880.
- Sivera Mascaró R., García Sobrino T., Horga Hernández A., et al. Clinical practice guidelines for the diagnosis and management of Charcot-Marie-Tooth disease. Neurologia (English Edition). 2025;40(3):290–305. Epub 01.03.2024.