Содержание
1. Описание, классификация и причины заболевания2. Симптоматика патологии3. Диагностические методы выявления4. Лечение и восстановительные меры5. Когда срочно к врачу?6. FAQ
Описание, классификация и причины заболевания
Патология имеет генетически выраженную природу, характеризуется врожденными нарушениями в строении мышечных структур организма. В зависимости от типа заболевания проявляется степень тяжести первичных и последующих признаков в клинической картине. Общая составляющая недуга состоит в явном тонусном снижении функций мышечной ткани, проявляющаяся в диффузной слабости.

Понятие «врожденная миопатия» включает целый ряд врожденных заболеваний, относящихся к категории редко встречающихся, но имеющих набор схожих симптомов и физиологических проявлений. Подразделяются они лишь по степени и локализации поражения мышечных волокон. Выявлена наследственная природа патологии, при этом дегенерирует ген, отвечающий за производство белков, которые является основополагающими в строение мышечного волокна. Результатом подобных изменений становится снижение сократительной способности мышечного аппарата и проявление его ослабленности. Как правило, заболевание становится заметно уже в раннем детстве, его признаки сохраняются на протяжении всей жизни человека. Чаще всего дегенеративные процессы протекают замедленно и без тяжелых проявлений, большинство пациентов вовсе не замечают явного прогрессирования аномалии.
За основу распределения заболевания по видам берется два блока информации:
- местоположение мутировавшего гена;
- тип белка, который претерпевает изменения.
В зависимости от полноты полученных исследовательским путем данных классифицируются подвиды врожденной миопатии:
- Немалиновая патология, деформация центрального стержня, миотобулярное поражение – данные болезни основываются на известных специалистам измененных генах и дефектных белковых соединениях.
- Десмин-связанная аномалия, актинзависимая миопатия – эти патологии определяются при наличии данных о мутировавшем гене, однако аномальный белок остается неизвестным.
- Диспропорция мышечных волокон, центронуклеарное поражение, патология с множественными стержнями – при постановке диагноза специалисты не могут ответить на вопрос, какой именно участок ДНК мутировал, и какой белок не поддается синтезу.
Симптоматика патологии
Заболевание проявляется уже с первых дней жизни ребенка. Такие дети отличаются вялостью, слабостью, недостаточной физической активностью, малым объемом мускульной структуры, трудно и слабо прикладываются к груди, так как им не хватает сил для качественного сосания. С ростом ребенка мышечная ослабленность становится более заметной. Ребенку трудно подняться, залезть на стул, ходить, переложить вещь с места на место и т.д. По степени тяжести имеют место вариации, но в большинстве случаев существенного прогресса атрофического процесса не происходит. При тяжелом поражении ребенок так и не может научиться ходить, что приковывает его к инвалидному креслу. Тем не менее те навыки, которые приобретаются человеком с детского возраста, на протяжении жизни не утрачиваются.
Существуют и косвенные признаки описываемого заболевания, проявляющиеся в физиологических, анатомических особенностях. У небольшого числа пациентов отмечается:
- изменение формы лицевой части – узкие, вытянутые черты черепа;
- высокое (в сравнении с нормой) небо;
- искривление позвоночного столба различной степени тяжести;
- кривоногость;
- врожденный вывих бедра.
Так как аномалия охватывает все мышечные структуры организма, страдает и мышечный аппарат дыхательной системы. Это опасно хроническим кислородным голоданием, у ребенка развивается дыхательная недостаточность. Это чревато возникновением таких сопутствующих нарушений, как часто повторяющийся бронхит, различные типы пневмонии. Дыхательная недостаточность особо опасна для грудных детей, так как может привести к внезапной остановке дыхания и смерти.
Если во время диагностики выявлена миопатия центрального стержня, помимо мускулатурой слабости главными признаками данной аномалии выступают:
- недостаточная мимика лица;
- невысокий рост, миниатюрность фигуры;
- явная деформация костей и позвоночника;
- наличие некротических вкраплений в забранной на анализ мышечной ткани.
Остальные виды миопатии характеризуются разной степенью проявления следующей симптоматики:
- недостаточная двигательная активность во внутриутробном периоде развития;
- синдром вялого ребенка;
- синдром ригидного позвоночника;
- расстройство глотательной и дыхательной функций;
- атрофия мимических мышц лицевой части.
Диагностические методы выявления
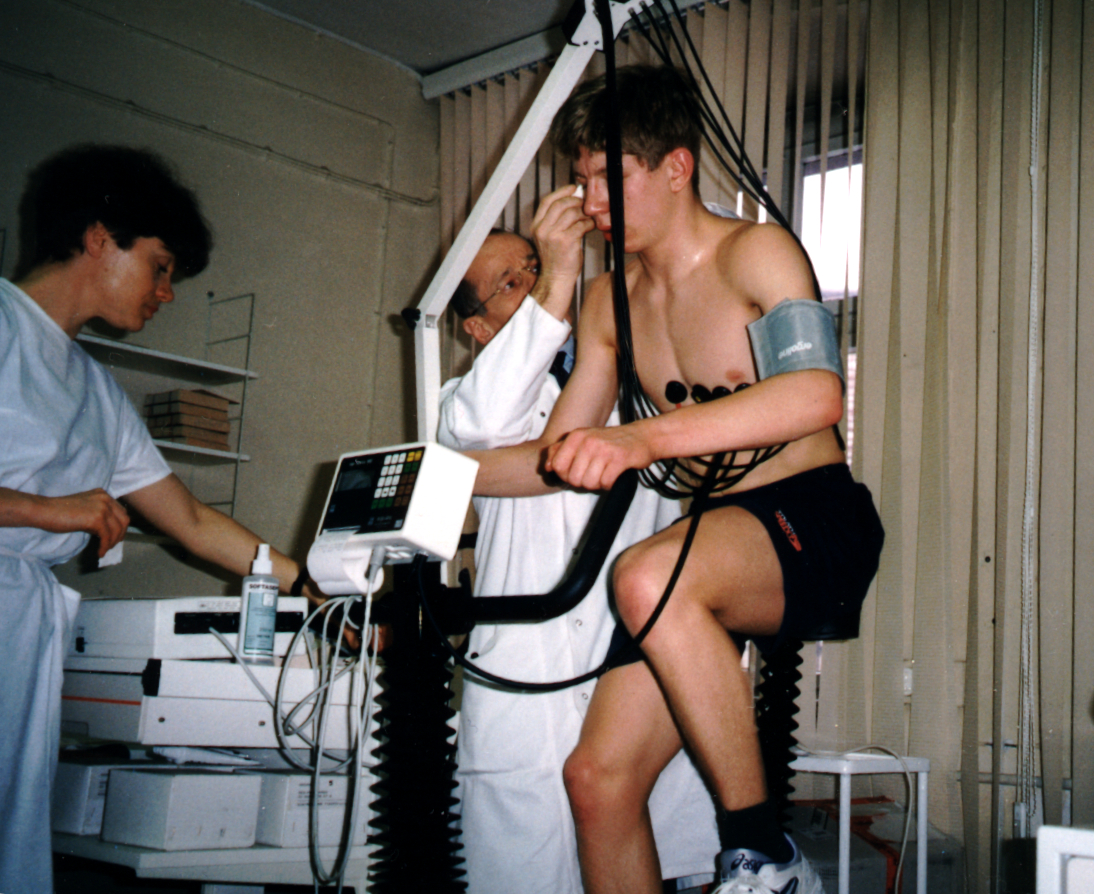
Так как основная симптоматика становится заметной уже с первых дней жизни (реже на более поздних этапах развития, еще реже в юношестве), заподозрить наличие такого заболевания, как врожденная миопатия, удается при первом же осмотре у невролога. Специалист проводит первичное тестирование по замерам силы и тонуса мышечного аппарата – эргометрию. Для сбора дополнительных данных для анамнеза применяются такие инструментальные методики, как электромиография, миотометрия и электротонометрия.
Для постановки итогового диагноза врачу необходимо исключить похожие по симптоматике заболевания, вызванные воспалительной причиной, инфицированием и т.д. О развитии врожденной классической миопатии говорит тот факт, что у пациентов с таки диагнозом отсутствует болезненность в атрофированных мышцах, самочувствие остается удовлетворительным, нет признаков протекающего воспалительного процесса, отмечающегося отечностью, повышением местной температуры тела и пальпируемых уплотнений ткани.
Самым информативным способом определения патологии выступает анализирование полученных мускульных волокон в результате биопсии. Каждый тип заболевания меняет структуры ткани по-разному. Определив эти изменения, выявляется форма болезни, ставится окончательный диагноз. Имеют место случаи неспецифической деструктуризации мышц, что затрудняет постановку клинического заключения. Для дифференциации других заболеваний проводят МРТ позвоночника.
Лечение и восстановительные меры
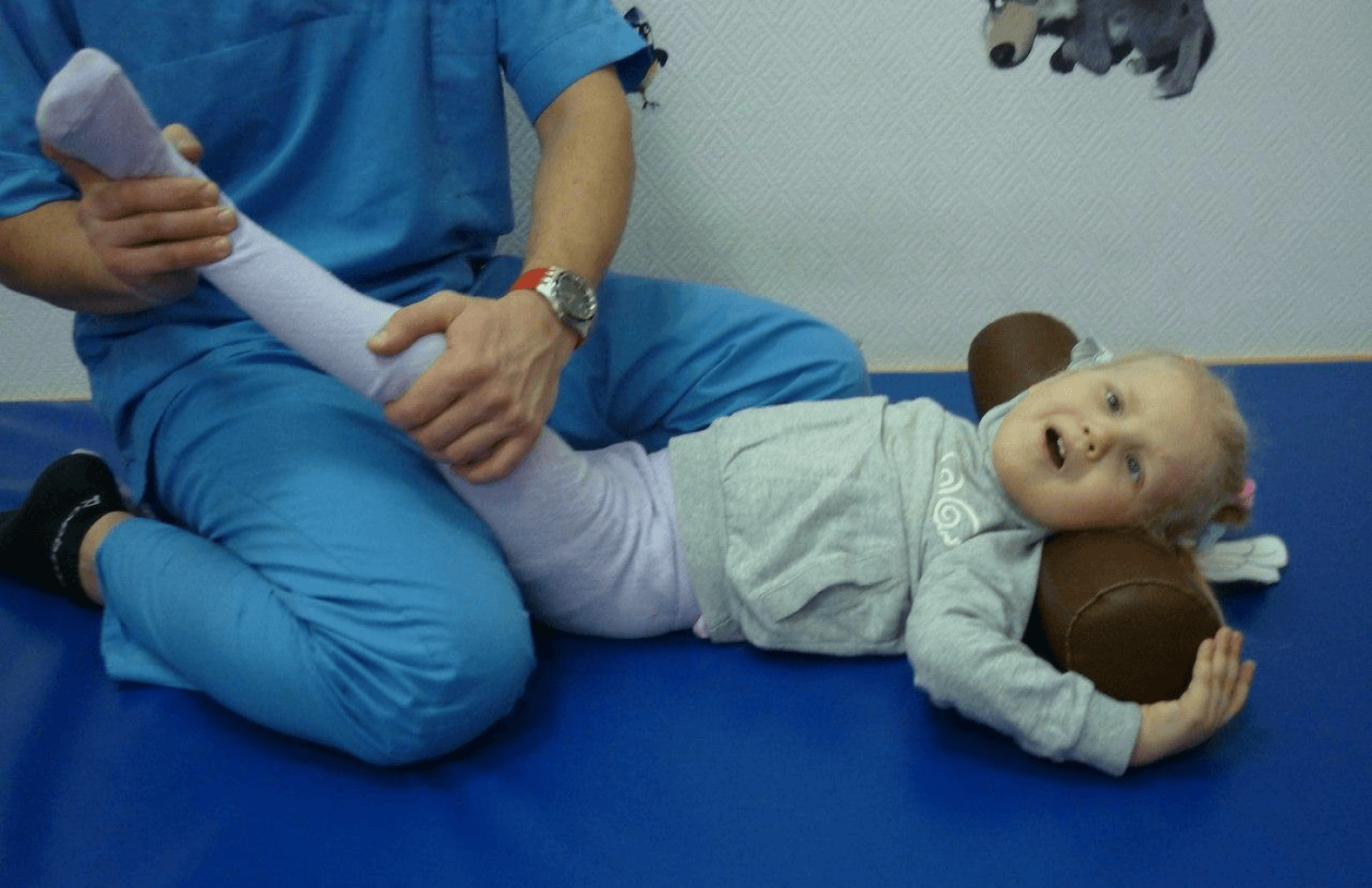
Так как рассматриваемое аномальное состояние является генетически обусловленным, заместить генерируемый собственным организмом белок не представляется возможным. Ни один из примененных ранее способов лечения не привел к положительным результатам. Симптоматика сохраняется на протяжении всей жизни больного. Лечебные манипуляции заключаются в поддерживающих процедурах, снижающих риск развития сопутствующих осложнений. Так, в младенчестве, если наблюдается сильное снижение тонуса, медики направляют все усилия на снижение дыхательной дисфункции. Параллельно нарушена и функция глотания, поэтому питательные растворы вводятся посредством зондирования. Проводится терапия бронхо-легочных патологий при первых признаках их появления.
Поддерживать удовлетворительное физическое состояние и качество жизни помогают такие процедуры, как систематические курсы массажа, терапия с помощью воды, посещение физиокабинета. С ростом возрастной категории пациента может понадобиться помощь ортопеда, который проведет возможную коррекцию. Если человек тяжело адаптируется в социуме на фоне своего заболевания, рекомендуется обратиться за психотерапевтической помощью, к общению в адаптационных группах.
Когда срочно к врачу?
Немедленно обратитесь за медицинской помощью, если у ребенка (или взрослого с диагностированной врожденной миопатией) появились следующие симптомы:
- Внезапное затруднение дыхания, одышка или остановка дыхания во сне.
- Сильная слабость, из-за которой ребенок перестал держать голову, сидеть или двигаться (утрата ранее приобретенных навыков).
- Проблемы с глотанием (поперхивание, невозможность проглотить пищу или воду).
- Посинение носогубного треугольника или бледность кожи.
- Частые и тяжелые приступы бронхита или пневмонии.
- Резкое искривление позвоночника, которое мешает сидеть или лежать.
FAQ
Что такое врожденная миопатия?
Это группа генетических заболеваний, при которых с рождения нарушено строение мышечных волокон. Из-за этого мышцы становятся слабыми и не могут нормально сокращаться. Болезнь обычно прогрессирует медленно или не прогрессирует вовсе, но сохраняется на всю жизнь.
Как проявляется болезнь у новорожденных?
Младенцы с миопатией часто вялые, мало двигаются, у них слабый крик и плохое сосание (быстро устают при кормлении). Может наблюдаться "синдром вялого ребенка", когда руки и ноги свисают как плети. Также возможны проблемы с дыханием из-за слабости дыхательных мышц.
Как точно диагностируют врожденную миопатию?
Самый точный метод — биопсия мышц (взятие маленького кусочка ткани для анализа под микроскопом), которая показывает характерные изменения структуры волокон. Также проводят электромиографию (оценку электрической активности мышц) и генетические тесты для поиска мутации.
Можно ли вылечить врожденную миопатию?
На сегодняшний день полное излечение невозможно, так как это генетическая патология. Однако поддерживающая терапия (массаж, ЛФК, физиопроцедуры, лечение дыхательных нарушений) помогает сохранить качество жизни, избежать осложнений и замедлить развитие слабости.
Как врожденная миопатия влияет на продолжительность жизни?
Многое зависит от формы и тяжести заболевания. При легких формах люди доживают до зрелого возраста и ведут относительно активный образ жизни. Основную опасность представляют дыхательная недостаточность и инфекции легких (пневмонии), поэтому важно своевременное наблюдение у врача и профилактика осложнений.
Используемая и рекомендуемая литература
- PubMed (обзор по диагностике): North K.N. et al. 2014 — https://pubmed.ncbi.nlm.nih.gov/24456932/. PubMed.
- CDC (официальные ICD-10-CM обновления 2021: введены отдельные коды для врождённых миопатий G71.22–G71.29): https://www.cdc.gov/nchs/icd/icd10cm.htm (см. FY2021 ICD-10-CM Addenda и Табличный список).
- WHO (ICD-10, рубрика G71.2 «Congenital myopathy»): https://icd.who.int/browse10/2019/en#/G71.2.
- Каплан Дж.Г. Версия таблицы генов моногенных нервно-мышечных заболеваний 2013 года. Нервно-мышечная. Раздор 2012.
- Wang C.H., Dowling J.J., North K., et al. Consensus statement on standard of care for congenital myopathies. Journal of Child Neurology. 2012;27(3):363–382. doi:10.1177/0883073812436605.
- Sewry C.A., Wallgren-Pettersson C. Myopathology in congenital myopathies. Neuropathology and Applied Neurobiology. 2017;43(1):5–23. doi:10.1111/nan.12369.
- Papadimas G.K., Papadopoulos C., et al. Update on Congenital Myopathies in Adulthood. International Journal of Molecular Sciences. 2020;21(10):3694. doi:10.3390/ijms21103694.